







我司抗原芯片首席技术官李博士受邀演讲|风湿免疫论坛直播
风湿免疫论坛直播
查看全部内容
2023-12-12



广州复能基因有限公司是以为人类基因功能研究和临床应用研究者提供实物和技术为宗旨的科技研发型企业。公司自成立以来和国际上一流研究机构、大型医药和生物技术企业进行广泛合作,参加了世界人类全长基因开放阅读框(ORF)协作组织。
成立时间
攻关课题
复能公司的克隆涵盖了几乎所有可能的分子生物学应用,包括ORF,CRISPR和TALEN,miRNA和启动子等。我们所有的ORF,shRNA,CRISPR sgRNA和TALEN克隆都经过序列验证,为您提供了最高的质量。我们大部分即用型表达克隆都在非病毒或慢病毒载体中,方便您在多种细胞类型中进行基因表达。
查看详细内容 
基因组编辑以其高效率、易用性和相对低廉的成本,为生物医学研究带来革命性的变革。复能公司的基因组编辑的产品有CRISPR-Cas9克隆、验证、筛选,预制的稳定细胞系,等等,为您提供基因组编辑完整的解决方案,在您基因组编辑工作流程的每一步提供帮助。
查看详细内容 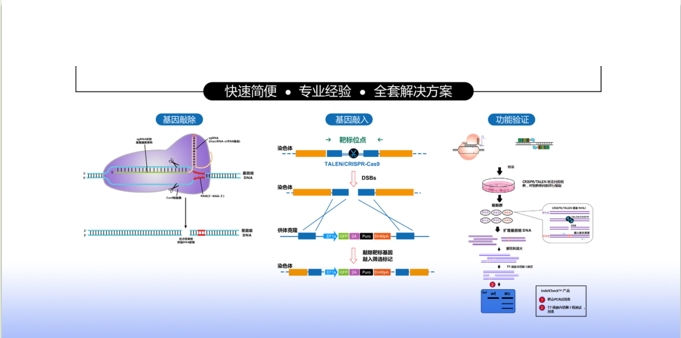
近来研究发现miRNA几乎与所有细胞的功能和生物体生命过程有关。MicroRNA所调节基因的表达失调与癌症、心血管疾病等各种疾病有关。复能基因提供microRNA (miRNA)完整研究方案,帮助客户检测、验证或敲低miRNA的表达,覆盖miRBase中人、小鼠、大鼠 miRNA。
查看详细内容 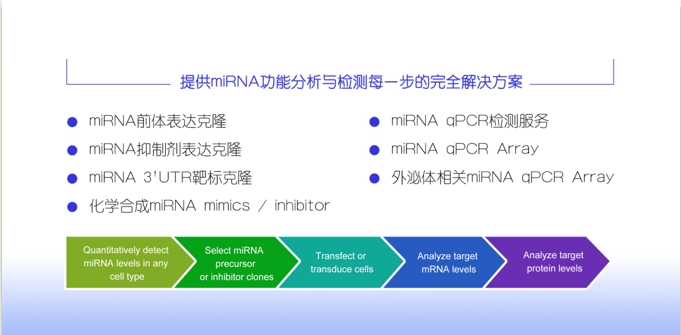
致力于成为向生物学和生物医学研究领域的研究创新和产品开发提供新技术、新产品,以及专有的工具和服务的主要供应商。 公司主要生产和销售与基因组学和蛋白质组学相关的、高质量的生物研究试剂和产品,包括核酸、蛋白质和抗体相关的产品和服务。

